Genomic Analysis of Intellectual Disability
Introduction
The interaction between the alleles and loci within the genome is included in genomic study. Akbani et al. (2015) opined that genomic is an area of study within the genetics that involves sequencing and analysis of the genome of an organism. The aim of the assignment is to study the given case study and critically analysis is based on the genomic study. The case is related to Roger, a 15 years who have a mild to moderate intellectual disability. His family history was also provided that would help in understanding the genome sequencing. For those seeking additional support, resources such as healthcare dissertation help can be invaluable in navigating complex genetic topics.
Part A
Critical discussion of key information and its significance
As stated by the case study, Roger was suffering from intellectual disability, which is a general learning disability and mental retardation. A neurodevelopment disorder is characterised by impairment of the intellectual as well as adaptive functioning. As per the opinion of Mouneshachari et al., (2016), the intelligent quotient of such people is under 70 and there is a deficit in two or more adaptive behaviours that affect the daily routine of the person. The disorder is apparently found during childhood and observed to have a number of defects in term of mental disabilities, social behaviours and activities within the daily living in comparison to others of similar age. In case of mild to moderate form of intellectual disability, there are no observable physical signs however, characteristic physical traits are associated with the disorder such as Down syndrome. The level of the disorder determines the severity of impairment in each individual. The early signs that help in the identification of the genetic disorder are failure or delay in reaching out or achieving milestones due to impairment in motor skills development, behavioural and social issue, slow in talking and continued issue with speech and language, poor planning abilities and lack of problem-solving abilities. Along with this, other signs include problems in school, issue with self-care and self-help, adaptation issue, Intellectual difficulties and problem in understanding and following rules (Balogh et al. 2016). Roger’s mother was healthy however, his father suffered from pancreatic cancer that led to his death. According to the medical terms, pancreatic cancer initiated from the tissue of the pancreas, which is an organ in the abdomen that lies in a horizontal position in the lower part of the stomach. The pancreas is responsible for releasing an enzyme that helps in digestion and hormone that regulates the blood sugar level (Zhou & Melton, 2018). This type of cancer spread rapidly to the adjoining organ and is rarely detected at the early stages. In some case, the detection of pancreatic cysts or family history may lead to screening of the problem at an early phase. One of the crucial sign of pancreatic cancer is diabetes that led to weight loss, pain in the upper abdomen as well as in the back and jaundice (Mayoclinic.org, 2019). The signs of pancreatic cancer that can be found only during the advanced stages include loss of appetite that leads to unintended weight loss, feeling fatigue, pain in the back and upper abdomen, blood clots in case of injury, early-onset of diabetes and yellowing of skin and whiteness in the eye, indicating jaundice.
Roger’s sister is married to a cousin (maternal uncle’s son) and they have two small girls. The younger one is six months old and has been born with an extra finger in each of her hand. This condition is known as polydactyly where a person is born with one or more extra finger or toes and can occur in one or both hands or feet (Mantilla-Rivas et al., 2019). The family may pass a genetic disease. When it is being passed by the family, it is known as familial polydactyly which occurs in isolation that is, the person does not have any symptoms, In case, it is not passed down, it occurs due to mutation that has occurred when the baby was in the womb. The symptoms that are associated with the condition may vary in nature and include small lumps of raised tissue that do not have any bone, partially formed finger or toes that may contain some bone but not joints and in some cases, they have fully functionally fingers and toes that have bone, tissue and joints (Al‐Qattan et al. 2017).
Diagrammatic representation of the family tree of Roger and its significance
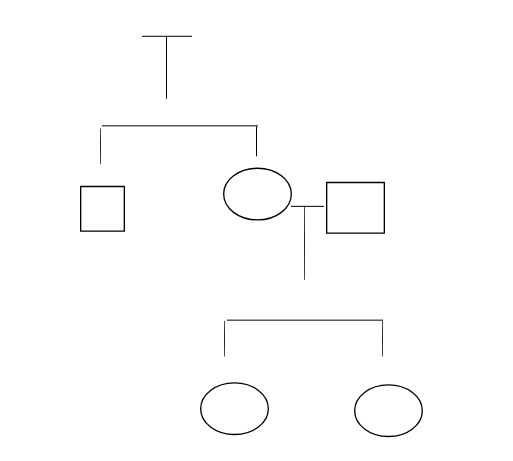
According to Westberg (2016), a pedigree chart is a diagram that represents the appearance and occurrence of phenotypes of a particular gene or organisms and its family from one generation to another and most commonly done in human, dogs and race horses. The results of this chart demonstrate the information about the family in the form of easily representative charts. The use of symbols where squares demonstrated males and circles are females. The construction of pedigree is a family history and details about the previous generation that is uncertain and fades with time (Rivera, 2016). From the above figure 1, the first generation is Roger’s father and mother where his mother is a healthy individual whereas his father had died due to pancreatic cancer. They have two children, Roger and his sister who is a healthy individual as per the information. His sister married to maternal uncle’s son and they have two daughters. The younger daughter who is six months old has a polydactyly that has an extra finger in each of the hands.
Critical discussion of likely mode of inheritance of Roger’s disorder
The inheritance of the disorder suffered by Roger is genetic in nature. As per the viewpoint of Vissers et al. (2016), it can be found that it is a neurodevelopment disorder that affects the nervous system that leading to abnormal brain function. This is likely to occur through two modes, environmental and genetic in nature. The deviation from the developmental trajectory in life may result in missing or abnormal neuronal connectivity. It can be stated that due to spatial or temporal complexities of the trajectory, there are many causes of the disorders that affect different parts of the nervous system at different times of an individual. In this particular case, the chance of mutation from father is a possibility (Livingston & Happé, 2017). The mutation occurred when the pancreas was affected due to external or internal outcomes. The mating of a mutated and healthy individual lead to the transfer of mutated alleles, which was dominant in Roger’s case as a result of which, the disorder was prevalent. In other cases, mutation may have occurred due to environmental factors that caused the disorder.
Critical explanation of application of genomic sequencing in this case
According to Lelieveld et al. (2016), the development disabilities have a number of a genetic cause that needs to be identified for proper diagnosis. The use of genome sequencing is found to be useful in this case. From the case as well as from the pedigree chart, data will be collected that will specifically highlight the case of Roger’s which was identified to be genetic in nature. It was reported by his mother that he had difficulty in walking up to three years of age and was diagnosed to have oculomotor apraxia. The sequencing of the genome is a critical step in understanding it in a complete manner. The genome sequence demonstrates a valuable shortcut and helps to find the faulty gene in an easy and quick manner. The genome sequence does not offer have a clue about the location of the gene however; it helps in interpreting the clues in a systematic manner. The study if the genome sequence aid in understanding the work of the genome and ways genes are working together in term of growth, development and maintenance of the organism or case. As per the study by Lelieveld et al. (2016), 25% of DNA accounts for the entire gene and this, understanding it allows in the study of parts outside the genes, which includes regulatory regions that are responsible for controlling the genes by turning them on and off as well as understanding the junk DNA present in the body.
Part B
Upon investigation of the human genome sequencing of Roger, three variants were identified and thus, critical analysis of each variant will be conducted based on the given criteria.
Critical assessments of all three variants
• Variant 1 Location (GRCh37): chr12:88483128C>T
Location (GRCh38): chr12:88089351C>T
Gene: CEP290
Variant: homozygous missense variant
a. Molecular biological consequence of the variants
The gene that was found in the variant 1 is known to be homozygous missense in nature. This indicates that fact that the gene has two identical alleles on the homologous chromosomes and due to point mutation, there is a change in one amino acid within a single nucleotide that causes nonsynonymous substitution in the DNA sequence. The gene is question CEP290 that is responsible for producing protein in many cells within the body (Spiegel et al. 2016). Even though the function of the protein is not completely understood however, studies indicates that it play a critical role in cell structures, cilia and centrosomes. The function of centrosomes is in cell division and thus, help in assembly of microtubules, which are the proteins that help in transporting of materials within the cells and help in maintaining the shape of the cell. On the other hand, cilia are finger-like microscopic projections that stick out of the surface of the cells. These are involved in cell movement and in chemical signalling pathways. Thus, these are important in perceiving the sensory inputs such as vision, hearing and smell (Zahid et al., 2018). The mutation in both the alleles that lead to impairment in the neurodevelopment system or Central Nervous System and the processing of the cells, leading to Intellectual Disability as well as Sensory difficulties among individual.
b. Allele frequency
As stated in the result of genome sequencing that variant 1 is homozygous in nature. This is indicates the fact that the gene has two identical alleles on the homologous chromosomes.
c. Likely pathogenicity
The health condition that may occur due to the genetic changes in the case are Bardet-Biedl syndrome, Leber congenital amaurosis, Meckel syndrome, Joubert syndrome, Senior-Loken syndrome and other diseases.
d. Diagnosis and relevance to Roger’s phenotype
It is evident that the cause of Roger’s phenotype is implied to the fact that it is due to genetic. The suspicion of the disorder can be done due to many reasons. In the event that an infant has physical anomalies that propose a hereditary or metabolic issue, an assortment of tests might be done to affirm the conclusion. These incorporate blood tests, urine tests, imaging tests to search for auxiliary issues in the cerebrum, or electroencephalogram (EEG) to search for proof of seizures (Fritz et al. 2016). In youngsters with formative deferrals, the specialist will perform tests to preclude different issues, including hearing issues and certain neurological issue (Crnic et al., 2017). On the off chance that no other reason can be found for the postponements, the kid will be alluded for formal testing.
e. Ethical implications for family members
Individuals with ID have, truth be told, assumed a critical job in the very development of research morals as a field of request. Huge numbers of the codes and arrangements administering research on human subjects that have been explained since the mid-twentieth century are an immediate reaction to the maltreatment and abuse endured by individuals with handicaps because of specialists.
f. Further testing or treatment implications for Roger and other members of the family
Genome sequencing of the family members can be conducted.
• Variant 2 Location (GRCh37): chr5:37167148C>T
Location (GRCh38): chr5:37167046C>T
Gene: CSORF42
Variant: homozygous splice donor variant
a. Molecular biological consequence of the variants
A graft site transformation is a hereditary change that supplements, erases or changes various nucleotides in the particular site at which joining happens during the handling of antecedent ambassador RNA into develop flag-bearer RNA. Join site accord successions that drive exon acknowledgment are situated at the very ends of introns. The erasure of the grafting site brings about at least one introns staying in develop mRNA and may prompt the creation of irregular proteins. At the point when a join site change happens, the mRNA transcript has data from these introns that typically ought not to be incorporated (Carlosama et al., 2017). Introns should be expelled, while the exons are communicated. The change must happen at the particular site at which intron grafting happens: inside non-coding destinations in a quality, legitimately beside the area of the exon. The transformation can be an inclusion, erasure, outline move, and so on. The joining procedure itself is constrained by the given successions, known as graft giver and join acceptor groupings, which encompass every exon. Changes in these arrangements may prompt maintenance of huge fragments of intronic DNA by the mRNA, or to whole exons being grafted out of the mRNA (Bakshi et al. 2018).
b. Allele frequency
Both the alleles are associated.
c. Likely pathogenicity
Because of the touchy area of join locales, changes in the acceptor or contributor territories of graft destinations can get hindering to a human person. Truth be told, a wide range of sorts of infections originate from irregularities inside the join destinations. The disease include Cancer, Dementia, Hematological Disorders, Parathyroid Deficiency and Epilepsy.
d. Diagnosis and relevance to Roger’s phenotype
Three factors into the finding of scholarly inability: interviews with the guardians, perception of the kid, and testing of insight and versatile practices. A youngster is viewed as mentally incapacitated on the off chance that the person in question has deficiencies in both IQ and versatile practices. In the event that just either is available, the youngster is not viewed as mentally impaired.
e. Ethical implications for family members
The interest of individuals with ID in biomedical research proceeds with today. A review of current clinical preliminaries including individuals with ID on the National Institutes of Health library of clinical preliminaries uncovers several studies including subjects with an assortment of conditions, extending from wide assignments like "mental hindrance" and "scholarly incapacity" to explicit conditions like Down disorder and Fragile X disorder (Richards et al. 2017).
f. Further testing or treatment implications for Roger and other members of the family
After a determination of scholarly handicap is made, a group of experts will survey the youngster's specific qualities and shortcomings. This encourages them decide how much and what rather help the kid should prevail at home, in school, and in the network.
• Variant 3 Location (GRCh37):
chr13:32945142_32945143delAG Location
(GRCh38): chr13:32371005_32371006delAG
Gene: BRCA2
Variant: heterozygous 2 base pair deletion
a. Molecular biological consequence of the variants
A frameshift change is not equivalent to a solitary nucleotide polymorphism. Those with a heterozygous transformation for the CCR5 were less defenceless to ... 8 of the watched transformations are frameshift, 6 cancellations and 2 inclusions.
b. Allele frequency
In this case, there is loss of one allele occur that the allele frequency is 1. Frameshift transformations are seen as increasingly normal in rehash areas of DNA. An explanation behind this is a direct result of slipping of the polymerase compound in rehash locales, taking into consideration transformations to enter the sequence. Experiments can be hurried to decide the recurrence of the frameshift change by including or expelling a pre-set number of nucleotides. Investigations have been controlled by including four basepairs, called the +4 tests, yet a group from Emory University took a gander at the distinction in recurrence of the transformation by both including and erasing a base pair (Yoshida et al., 2016). It was demonstrated that there was no distinction in the recurrence between the expansion and cancellation of a base pair. There is be that as it may, a distinction at last consequence of the protein.
c. Likely pathogenicity
Frameshift changes can happen arbitrarily or be brought about by an outside boost. The discovery of frameshift changes can happen through a few unique techniques. Frameshifts are only one kind of transformation that can prompt fragmented or off base proteins, however they represent a noteworthy level of blunders in DNA. This is a hereditary transformation at the degree of nucleotide bases. Why and how frameshift changes happen are consistently being looked for after. An ecological examination, explicitly the generation of UV-instigated frameshift transformations by DNA polymerases lacking in 3′ → 5′ exonuclease movement was finished (Kuzminov, 2019) . The ordinary arrangement 5′ GTC GTT TTA CAA 3′ was changed to GTC GTT T TTA CAA (MIDT) of GTC GTT C TTA CAA (MIDC) to examine frameshifts. E. coli pol I Kf and T7 DNA polymerase freak chemicals without 3′ → 5′ exonuclease movement produce UV-prompted revertants at higher recurrence than did their exonuclease capable partners. The information demonstrates that loss of editing movement builds the recurrence of UV-initiated frameshifts.
d. Diagnosis and relevance to Roger’s phenotype
Subtleties the techniques and reagents for analysis of ailments brought about by or related with a quality having a substantial transformation offering ascend to a frameshift change. The strategies incorporate giving a tissue or liquid example and directing quality investigation for frameshift transformation or a protein from this sort of change. The nucleotide succession of the speculated quality is given from distributed quality groupings or from cloning and sequencing of the presume quality. The amino corrosive arrangement encoded by the quality is then anticipated.
e. Ethical implications for family members
These examinations, which incorporate medication preliminaries, observational investigations, hereditary research, and social research, center around both the nature and etiology of the incapacity itself and on related wellbeing conditions (e.g., Alzheimer's, malignant growth, metabolic infections, weight, mental issue.)
f. Further testing or treatment implications for Roger and other members of the family
Maybe the most outstanding case, following on the impact points of the revulsions of Nazi prescription, is the hepatitis study led at the Willowbrook State School for kids with mental hindrance in Staten Island, New York. Through the span of very nearly 2 decades, analysts purposely contaminated youngsters with the live hepatitis infection to examine the impacts of gamma globulin to treat it, damaging various moral standards.

Conclusion
Genomic is the study of the intragenomic process involves that include heterosis, epistasis and pleiotrophy. Genome sequencing is a process that helps in figuring the order of DNA bases or nucleotides within a genome that are of the order of Ac, Gs, Cs and Ts, the building blocks of the organism. There are over three billion genetic letters makes up the human genome.
Reference List
Akbani, R., Akdemir, K.C., Aksoy, B.A., Albert, M., Ally, A., Amin, S.B., Arachchi, H., Arora, A., Auman, J.T., Ayala, B. and Baboud, J., 2015. Genomic classification of cutaneous melanoma. Cell, 161(7), pp.1681-1696.
Al‐Qattan, M. M., Shamseldin, H. E., Salih, M. A., and Alkuraya, F. S. 2017. GLI3‐related polydactyly: a review. Clinical genetics, 92(5), pp.457-466.
Bakshi, A., Bretz, C. L., Cain, T. L., and Kim, J. 2018. Intergenic and intronic DNA hypomethylated regions as putative regulators of imprinted domains. Epigenomics, 10(4), pp.445-461.
Balogh, R., McMorris, C. A., Lunsky, Y., Ouellette‐Kuntz, H., Bourne, L., Colantonio, A., and Gonçalves‐Bradley, D. C. 2016. Organising healthcare services for persons with an intellectual disability. Cochrane Database of Systematic Reviews, (4).
Carlosama, C., Elzaiat, M., Patiño, L.C., Mateus, H.E., Veitia, R.A. and Laissue, P., 2017. A homozygous donor splice-site mutation in the meiotic gene MSH4 causes primary ovarian insufficiency. Human molecular genetics, 26(16), pp.3161-3166.
Crnic, K.A., Neece, C.L., McIntyre, L.L., Blacher, J. and Baker, B.L., 2017. Intellectual disability and developmental risk: Promoting intervention to improve child and family well‐being. Child Development, 88(2), pp.436-445.
Fritz, B. A., Kalarickal, P. L., Maybrier, H. R., Muench, M. R., Dearth, D., Chen, Y., ... and Avidan, M. S. 2016. Intraoperative electroencephalogram suppression predicts postoperative delirium. Anesthesia and analgesia, 122(1), p.234.
Lelieveld, S.H., Reijnders, M.R., Pfundt, R., Yntema, H.G., Kamsteeg, E.J., de Vries, P., de Vries, B.B., Willemsen, M.H., Kleefstra, T., Löhner, K. and Vreeburg, M., 2016. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nature neuroscience, 19(9), p.1194.
Livingston, L. A., and Happé, F. 2017. Conceptualising compensation in neurodevelopmental disorders: Reflections from autism spectrum disorder. Neuroscience & Biobehavioral Reviews, 80, pp.729-742.
Mantilla-Rivas, E., Tan, P., Zajac, J., Tilt, A., Rogers, G. F., and Oh, A. K. 2019. Is Epinephrine Safe for Infant Digit Excision? A Retrospective Review of 402 Polydactyly Excisions in Patients Younger than 6 Months. Plastic and reconstructive surgery, 144(1), pp. 149-154.
Mouneshachari, S., Pande, M.S. and Rao, T.S., 2016, February. EQ and IQ based classification of intelligent index (S-quotient) using K-means. In 2016 IEEE 6th International Conference on Advanced Computing (IACC) (pp. 101-105). IEEE.
Richards, C., Powis, L., Moss, J., Stinton, C., Nelson, L., and Oliver, C. 2017. Prospective study of autism phenomenology and the behavioural phenotype of Phelan–McDermid syndrome: comparison to fragile X syndrome, Down syndrome and idiopathic autism spectrum disorder. Journal of neurodevelopmental disorders, 9(1), 37.
Spiegel, R., Soiferman, D., Shaag, A., Shalev, S., Elpeleg, O., and Saada, A. 2016. Novel homozygous missense mutation in SPG20 gene results in Troyer syndrome associated with mitochondrial cytochrome c oxidase deficiency. In JIMD Reports, Volume 33 (pp. 55-60). Springer, Berlin, Heidelberg.
Yoshida, N., Miyoshi, H., Kato, T., Sakata‐Yanagimoto, M., Niino, D., Taniguchi, H., Moriuchi, Y., Miyahara, M., Kurita, D., Sasaki, Y. and Shimono, J., 2016. CCR4 frameshift mutation identifies a distinct group of adult T cell leukaemia/lymphoma with poor prognosis. The Journal of pathology, 238(5), pp.621-626.
Zahid, S., Branham, K., Schlegel, D., Pennesi, M.E., Michaelides, M., Heckenlively, J. and Jayasundera, T., 2018. CEP290. In Retinal Dystrophy Gene Atlas (pp. 47-49). Springer, Cham.



- 24/7 Customer Support
- 100% Customer Satisfaction
- No Privacy Violation
- Quick Services
- Subject Experts